南湖新闻网讯(通讯员 施雪萍)近日,我校园艺植物生物学教育部重点实验室郑波教授课题组在Briefings in Bioinformatics在线发表了题为“CProtMEDIAS: clustering of amino acid sequences encoded by gene families by MErging and DIgitizing Aligned Sequences”的研究论文。该论文通过蛋白序列位点特征进行分数矩阵构建及其降维分析,从而推断基因家族间的聚类和进化关系,在保证序列聚类稳定性的情况下,有效提高了序列聚类速度,为蛋白系统发育分析、发育轨迹分析提供了新的工具和方法。蛋白家族序列位点特征和降维分析的使用是该研究的主要创新点。
蛋白家族系统发育分析基于同源蛋白多序列比对推断蛋白间的进化关系,为蛋白家族的鉴定、蛋白功能和进化分析、基因组比较、物种鉴定等提供重要依据。基于同源蛋白多序列比对结果,通常有两类对其进行系统发育分析的方法,分别基于序列位点特征(包括极大似然法、最大简约法和贝叶斯推断法)和基于序列间进化距离(包括邻接法、最小进化法和非加权组平均法)。相比于基于序列间进化距离的分析方法,基于序列位点特征的方法能保留更多的序列信息,因此能获得更准确的结果。但由于极大似然法、贝叶斯推断法计算量过大,而最大简约法只适用于近缘序列,其适用性不如邻接法广泛。最小进化法在邻接法的基础上搜索分支长度最小的树,本质与邻接法相同。非加权组平均法假设碱基或氨基酸的替换速率是均等且恒等的,不利于模拟氨基酸序列之间的进化关系。
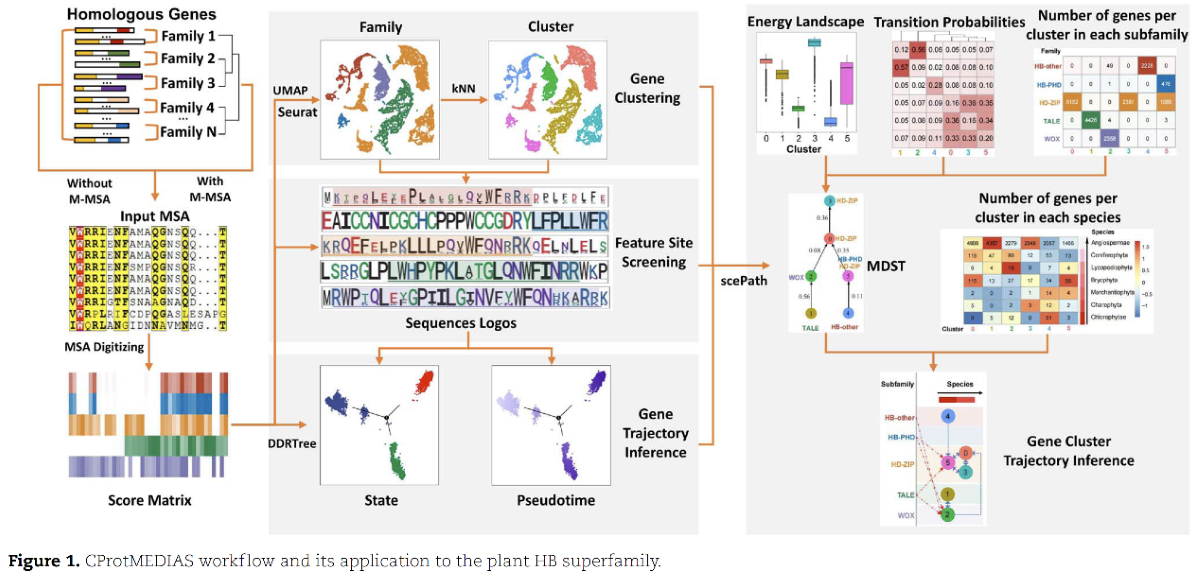
为解决上述问题,作者通过(1)合并多个蛋白家族或亚家族的多序列比对结果(Multiple Sequence Alignment,MSA);(2)使用合并后的MSA构建分数矩阵;(3)使用氨基酸序列比对融合方法UMAP和树形判别降维算法DDRTree对分数矩阵进行降维,之后使用共享最近邻算法SNN对输入序列进行聚类;(4)鉴定每一类输入序列保守位点及特异位点;(5)对分数矩阵进行拟时间分析;(6)推断各家族输入序列的发育轨迹等步骤,开发了一个用户友好的蛋白序列系统发育分析R语言软件包CProtMEDIAS,并以研究广泛、分析层次多、家族分类清晰的植物同源异型盒(Homeobox,HB)蛋白超家族为例,展示了CProtMEDIAS的分析流程,对该方法的有效性和准确性进行了评估。CProtMEDIAS具有适用于远缘序列、聚类稳定性好、分析速度快和图片精美等优点。
我校园艺林学学院张哲博士为该论文的第一作者,施雪萍副教授和郑波教授为该论文的共同通讯作者。该研究得到了国家自然科学基金的资助。
审核人:郑波
【英文摘要】
Protein phylogenetic analysis focuses on the evolutionary relationships among related protein sequences and can help researchers infer protein functions and developmental trajectories. With the advent of the big data era, the existing protein phylogenetic methods, including distance matrix and character-based methods, are facing challenges in both running time and application scope. Here, we developed an R package that we call CProtMEDIAS that is useful for protein phylogenetic analysis. In contrast to existing phylogenetic analysis methods, CProtMEDIAS utilizes dimensionality reduction algorithms to digitize multiple sequence alignments and quickly conduct phylogenetic analysis with a large number of amino acid sequences from similarly distant protein families and species. We used CProtMEDIAS to perform a dimensionality reduction, clustering, pseudotime, specific residue and evolutionary trajectory analysis of the plant homeobox superfamily. We found that CProtMEDIAS delivers consistent clustering, fast running and elegant presentation and thus provides powerful new tools and methods for protein clustering and evolutionary analysis.
原文链接:https://pubmed.ncbi.nlm.nih.gov/35834931/



 最近新闻
最近新闻