南湖新闻网讯(通讯员 王靖天)2024年6月18日,华中农业大学植物科学技术学院章元明教授团队在生物信息学领域重要刊物Briefings in Bioinformatics上发表了题为“FastBiCmrMLM: a fast and powerful compressed variance component mixed logistic model for big genomic case-control genome-wide association study”的研究论文,报道了全基因组关联分析(GWAS)方法学研究的重要进展。
该研究提出了一种多位点混合逻辑回归全模型的大型生物样本关联分析高效快速FastBiCmrMLM算法与软件,专门检测抗感二分类性状或case-control数据的性状与标记关联,是目前运行速度很快、检测功效最高和假阳性控制严格的二分类性状关联分析新工具。
GWAS是一种广泛应用于动植物、微生物和人类遗传学的基因挖掘方法。它通过检测标记与性状表型的关联,以挖掘控制复杂性状的基因,进而揭示复杂性状的遗传基础。以混合线性模型为基础的GWAS方法是在控制群体结构和遗传背景情况下高功效检测性状与标记间的关联,在过去20年得到广泛应用和发展。然而,目前的方法存在以下问题。第一,随着英国生物样本库(UK Biobank)等大型生物样本库的出现,关联群体样本量达到数十万甚至数百万,混合线性模型GWAS方法面临耗时、耗运算资源的挑战;第二,现有的大多GWAS方法通常只考虑等位基因替代效应及其遗传背景,导致估计的效应(混杂)和控制的多基因背景均不全面,且需要假定随机交配,降低检测功效与精度;最后,作物抗性性状关联分析通常采用连续型性状关联分析方法,也降低了位点检测功效与精度。
为解决上述问题,本研究创建了二分类性状的压缩方差组分混合逻辑回归全模型。该模型全面考虑了所有可能效应和遗传背景,采用章元明教授团队已建立的压缩方差组分技术(Li et al. 2022)将四个方差组分压缩为两个,显著降低了运算复杂度,结合了一系列快速算法和管理内存方法,发展了FastBiCmrMLM算法(图1),特别地,将SNP与性状关联推进至由连锁不平衡标记构建的bin或基因单倍型与性状关联,为作物抗性性状和人类复杂疾病基因挖掘提供新工具。
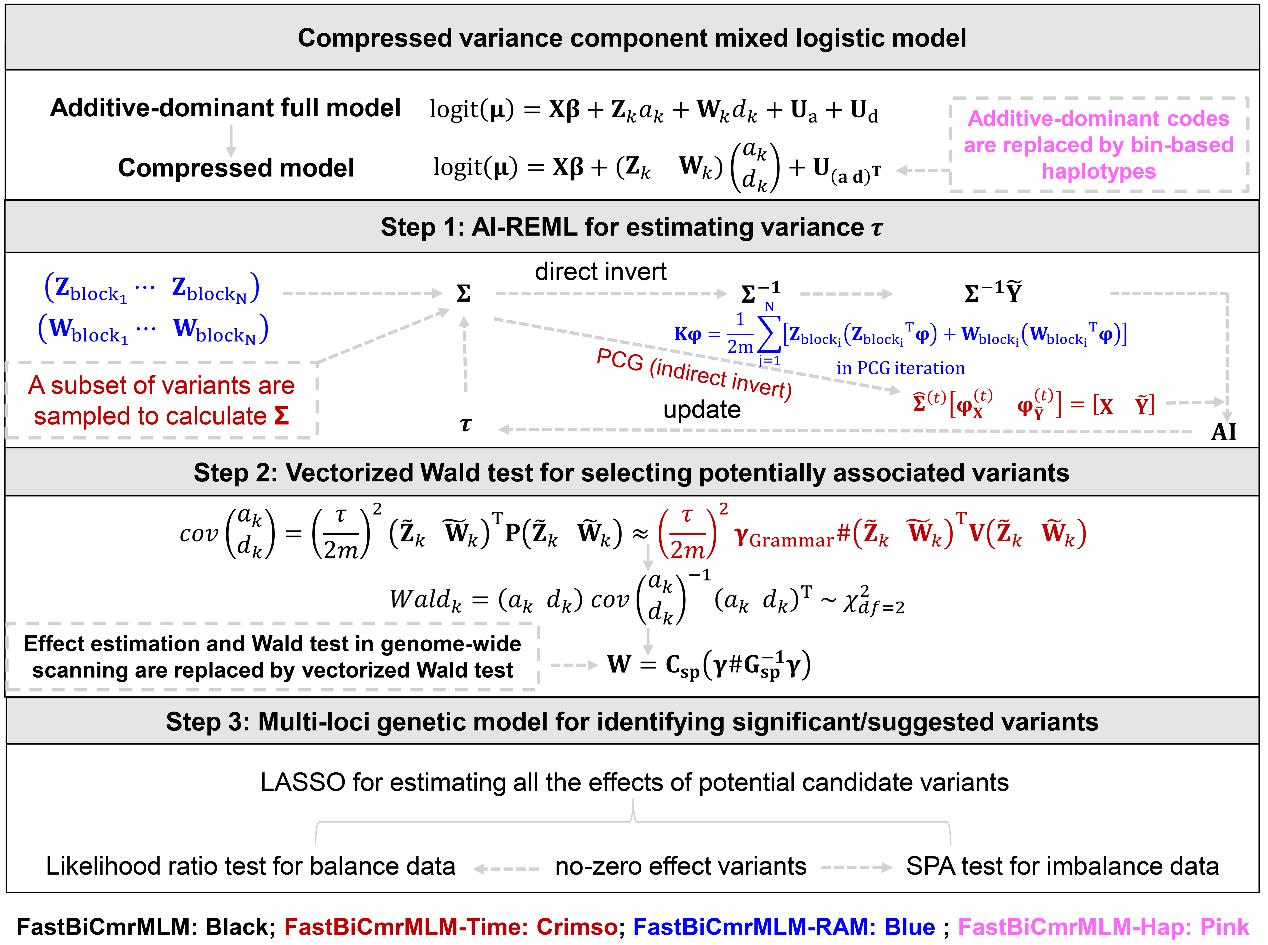
图1. FastBiCmrMLM算法的总体框架
FastBiCmrMLM新算法包含了4个模块以满足数据分析对样本容量、运算速度、节省内存和功能标记的需求。分析1000个体100万标记数据集大约需要7分钟;分析50万个体100万标记大型生物样本库规模的数据集大约需要14小时,且可检测大小为3‱、等位基因替代效应趋近于零和显性效应的位点。在模拟研究中,新算法的检测功效比现有二分类GWAS方法更高(图2);在5×10-8的显著概率阈值下,新算法假阳性率为4.2×10-8~4.8×10-8,很好地控制了假阳性率。此外,快速高效的新算法为多组学数据分析提供新工具。为检测到更多的基因,将关联分析从SNP标记分析推进到bin或基因单倍型分析,拓展了FastBiCmrMLM-Hap模块。该模块在模拟研究中可检测到频率为1.1%的稀有位点;在实际数据分析中能检测到更多的稀有(



 最近新闻
最近新闻