
南湖新闻网讯(通讯员 刘东旭)近日,我校生物信息团队杨庆勇课题组联合新加坡国立大学Sung Wing-Kin(宋永健)课题组研究成果以“Calling large indels in 1047 Arabidopsis with IndelEnsembler”为题在“Nucleic Acids Research”发表。研究开发出高性能的大片段插入删除变异(InDel)鉴定工具‘IndelEnsembler’,使大片段InDel鉴定准确性由45%提高到84%。
遗传变异是指一个群体中个体之间DNA序列的差异,主要包括点突变(SNP/单核苷酸多态性)、小片段InDel和结构变异(包括大片段InDel、染色体倒位、易位和拷贝数变异)。SNP被广泛应用于群体遗传学研究和疾病相关基因的研究。然后越来越多的研究表明,SNP并不能解释所有的表型差异,在人类中有超过1000种疾病由结构变异导致。
据介绍,大量的遗传学和分子生物学证据表明大片段InDel在解释影响一系列重要农作物的表型变异中起主要作用。例如,黄瓜基因组上一个大片段DNA序列拷贝数变化可以决定黄瓜的性别,包含该变异的黄瓜产量约为普通黄瓜的15倍。蟠桃因其果形独特,味甜多汁而受到人们喜爱,研究发现蟠桃基因组中含有一个1.67 Mb的倒位,而普通桃中则没有。但是,目前鉴定到的表型相关的大片段InDel的数量远远小于SNP,其主要原因是现有的方法不能精确的鉴定基因组上的大片段InDel。该研究通过整合四个已发表的方法开发出高性能的大片段InDel鉴定工具IndelEnsembler,并将其应用于由1047个拟南芥品系构成的自然群体中,通过全基因组关联分析(GWAS)鉴定到与重要性状相关的、新的大片段InDel。
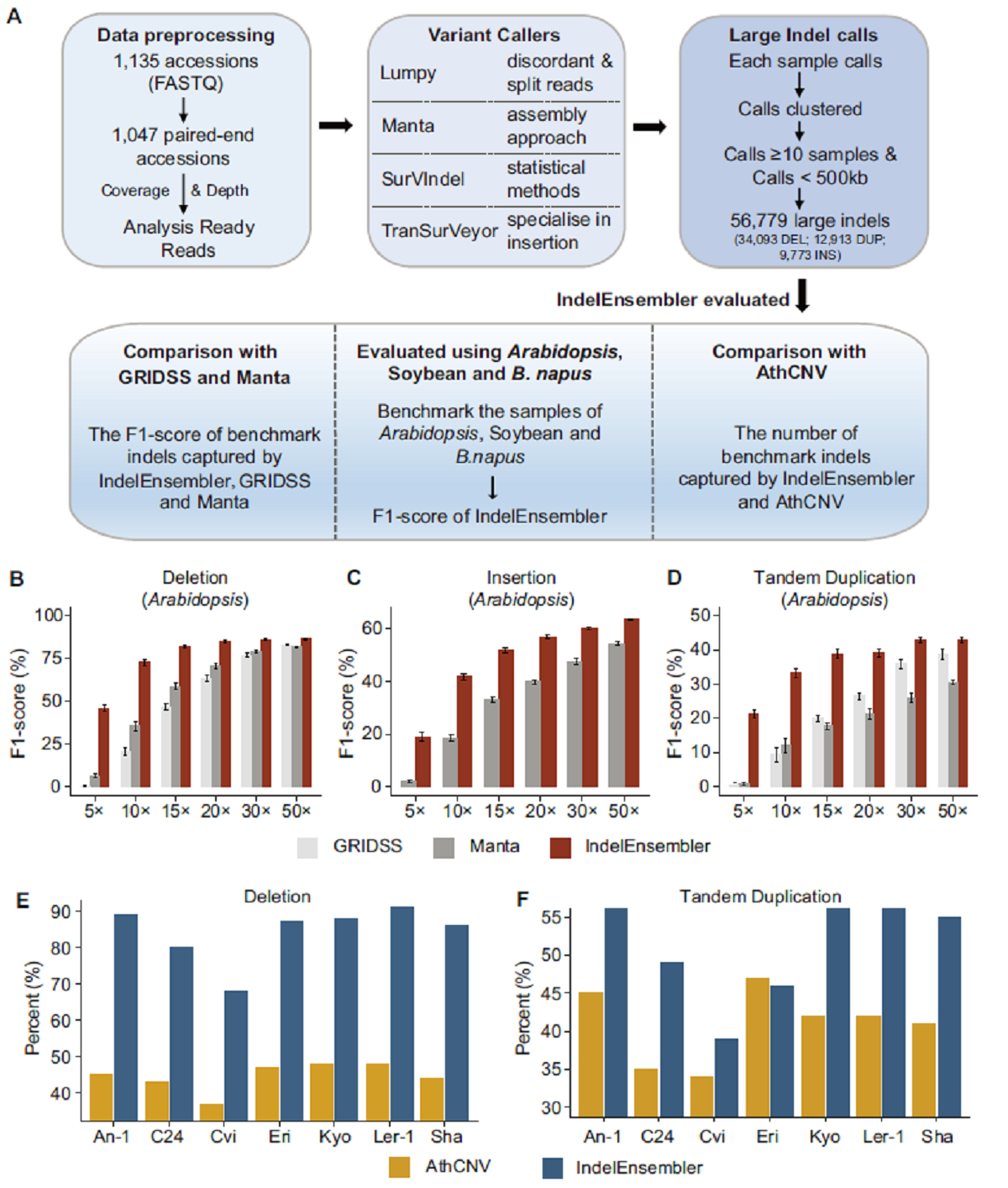
相比于目前鉴定InDel性能最佳的两个软件GRIDSS和Manta,IndelEnsembler在不同物种及不同测序深度下均有很好的性能。相比于拟南芥中已开发的工具AthCNV,IndelEnsembler在鉴定缺失变异(DEL)和重复变异(DUP)时准确性分别提高一倍和30%。
大片段InDel在拟南芥基因组上分布不均匀且与转座子的分布呈现极显著正相关,80-96%的染色体着丝粒区域被InDel覆盖。大片段InDel倾向于分布在基因间区以及非编码基因上。拟南芥基因组上有13,102个基因受到大片段InDel的影响,但是这些基因主要是未知功能的基因,说明大片段InDel影响的基因大部分为非核心基因。现有研究表明,非核心基因与水稻、玉米等重要农作物的适应性、品质和驯化等性状密切相关。此外,大片段InDel影响的基因更多的分布在基因组上的串联重复区域,说明基因组上的串联重复区域为不稳定区域会积累更多的变异。
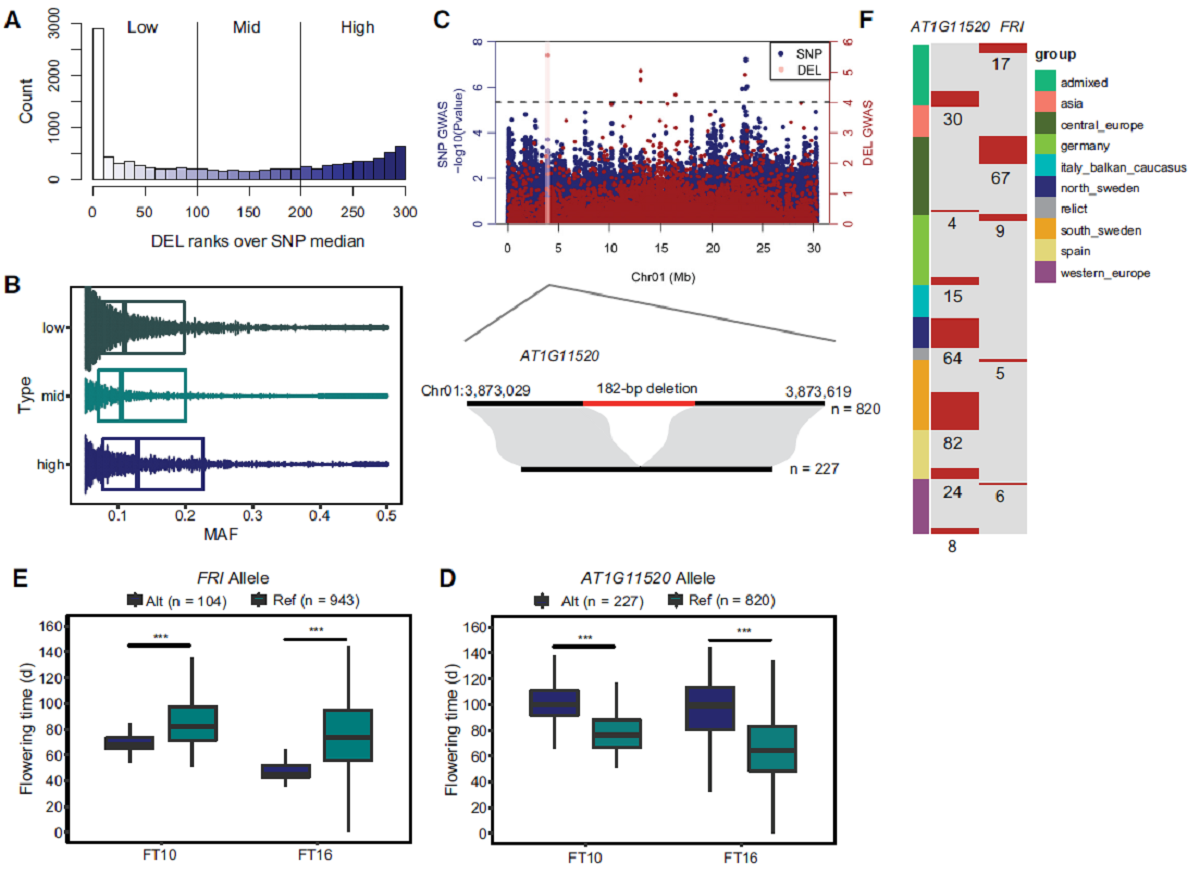
有48.91%的DEL与周围的SNP具有低连锁不平衡(LD),说明IndelEnsembler鉴定到了大量新的基因组变异,为挖掘影响拟南芥性状相关的基因组变异提供了重要资源。该研究鉴定到的一段377 bp的DEL造成开花期相关基因FRI第一个外显子上65 bp的缺失,含有该缺失的材料开花期提前。另外该研究还鉴定到AT1G11520基因上的一段182 bp缺失,含有该缺失的材料开花期推迟。值得注意的是,上述两段缺失不能在同一个材料中出现而且所有北瑞典地区的材料AT1G11520基因上均含有该182 bp的缺失。这些结果说明拟南芥中仍然存在不少未知的基因组变异,IndelEnsembler鉴定到的大片段InDel可以作为拟南芥中表型相关的基因组变异数据集的一个补充。
据悉我校信息学院博士研究生刘东旭和新加坡国立大学Ramesh Rajaby博士为该论文共同第一作者,信息学院杨庆勇副教授和新加坡国立大学宋永健教授为该论文通讯作者。该研究数据分析工作得到华中农业大学作物遗传改良国家重点实验室生物信息计算平台支持和帮助,得到国家重点研发计划(2017YFE0104800),国家自然科学基金(32070559)等项目资助。
审核人:杨庆勇
英文摘要
Large indels greatly impact the observable phenotypes in different organisms including plants and human. Hence, extracting large indels with high precision and sensitivity is important. Here, we developed IndelEnsembler to detect large indels in 1047 Arabidopsis whole-genome sequencing data. IndelEnsembler identified 34,093 deletions, 12,913 tandem duplications and 9,773 insertions. Our large indel dataset was more comprehensive and accurate compared with the previous dataset of AthCNV (1). We captured nearly twice of the ground truth deletions and on average 27% more ground truth duplications compared with AthCNV, though our dataset has less number of large indels compared with AthCNV. Our large indels were positively correlated with transposon elements across the Arabidopsis genome. The non-homologous recombination events were the major formation mechanism of deletions in Arabidopsis genome. The Neighbor joining (NJ) tree constructed based on IndelEnsembler’s deletions clearly divided the geographic subgroups of 1047 Arabidopsis. More importantly, our large indels represent a previously unassessed source of genetic variation. Approximately 49% of the deletions have low linkage disequilibrium (LD) with surrounding single nucleotide polymorphisms. Some of them could affect trait performance. For instance, using deletion-based genome-wide association study (DEL-GWAS), the accessions containing a 182-bp deletion in AT1G11520 had delayed flowering time and all accessions in north Sweden had the 182-bp deletion. We also found the accessions with 65-bp deletion in the first exon of AT4G00650 (FRI) flowered earlier than those without it. These two deletions cannot be detected in AthCNV and, interestingly, they do not co-occur in any Arabidopsis thaliana accession. By SNP-GWAS, surrounding SNPs of these two deletions do not correlate with flowering time. This example demonstrated that existing large indel datasets miss phenotypic variations and our large indel dataset filled in the gap.
论文链接:https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkab904/6395340